Retinoblastoma
Definition
Retinoblastoma is a malignant tumor of the retina that occurs predominantly in young children.
Description
The eye has three layers, the sclera, the choroid, and the retina. The sclera is the outer protective white coating of the eye. The choroid is the middle layer and contains blood vessels that nourish the eye. The front portion of the choroid is colored and is called the iris. The opening in the iris is called the pupil. The pupil is responsible for allowing light into the eye and usually appears black. When the pupil is exposed to bright light it contracts (closes), and when it is exposed to low light conditions it dilates (opens) so that the appropriate amount of light enters the eye. Light that enters through the pupil hits the lens of the eye. The lens then focuses the light onto the retina, the innermost of the three layers. The job of the retina is to transform the light into information that can be transmitted to the optic nerve, which will transmit this information to the brain. It is through this process that people are able to see the world around them.
Occasionally a tumor, called a retinoblastoma, develops in the retina of the eye. Usually this tumor forms in young children, but it can occasionally occur in adults. Most people with retinoblastoma develop only one tumor (unifocal) in only one eye (unilateral). Some, however, develop multiple tumors (multifocal) in one or both eyes. When retinoblastoma occurs independently in both eyes, it is then called bilateral retinoblastoma.
Occasionally, children with retinoblastoma develop trilateral retinoblastoma, which results from the development of an independent brain tumor that forms in a part of the brain called the pineal gland. In order for retinoblastoma to be classified as trilateral retinoblastoma, the tumor must have developed independently and not as the result of the spread of the retinal cancer . The prognosis for trilateral retinoblastoma is quite poor.
The retinal tumor which characterizes retinoblastoma is malignant, meaning that it can metastasize (spread) to other parts of the eye and eventually other parts of the body. In most cases, however, retinoblastoma is diagnosed before it spreads past the eye to other parts of the body (intraocular) and the prognosis is quite good. The prognosis is poorer if the cancer has spread beyond the eye (extraocular).
Retinoblastoma can be inherited or can arise spontaneously. Approximately 40 percent of people with retinoblastoma have an inherited form of the condition and approximately 60 percent have a sporadic (not inherited) form. Individuals with multiple independent tumors, bilateral retinoblastoma, or trilateral retinoblastoma are more likely to be affected with the inherited form of retinoblastoma.
Demographics
Approximately one in 15,000 to one in 30,000 infants are born with retinoblastoma, making it the most common childhood eye cancer. It is, however, a relatively rare childhood cancer and accounts for approximately 3 percent of childhood cancers. Retinoblastoma is found mainly in children under the age of five but can occasionally be seen in older children and adults. Retinoblastoma is found in individuals of all ethnic backgrounds and is found equally frequently in males and females.
Causes and symptoms
Causes
Retinoblastoma is caused by changes in or absence of a gene called RB1. RB1 is located on chromosome 13. Cells of the body, with the exception of the egg and sperm cells, contain 23 pairs of chromosomes. All of the cells of the body excluding the egg and the sperm cells are called the somatic cells. The somatic cells contain two of each chromosome 13 and, therefore, two copies of the RB1 gene. Each egg and sperm cell contains only one copy of chromosome and, therefore, only one copy of the RB1 gene.
RB1 produces a tumor suppressor protein that normally helps to regulate the cell cycle of cells such as those of the retina. A normal cell of the retina goes through a growth cycle during which it produces new cells. Genes such as tumor suppressor genes tightly regulate this growth cycle.
Cells that lose control of their cell cycle and replicate out of control are called cancer cells. These undergo many cell divisions, often at a quicker rate than normal cells and do not have a limited lifespan. A group of adjacent cancer cells can form a mass called a tumor. Malignant (cancerous) tumors can spread to other parts of the body. A malignant tumor of the retina (retinoblastoma) can result when just one retinal cell loses control of it cell cycle and replicates out of control.
Normally, the tumor suppressor protein produced by RB1 prevents a retinal cell from becoming cancerous. Each RB1 gene produces tumor suppressor protein. Only one functioning RB1 gene in a retinal cell is necessary to prevent the cell from becoming cancerous. If both RB1 genes in a retinal cell become non-functional, then a retinal cell can become cancerous and retinoblastoma can result. An RB1 gene is non-functional when it is changed or missing (deleted) and no longer produces normal tumor suppressor protein.
Approximately 40 percent of people with retinoblastoma have inherited a non-functional or deleted RB1 gene from either their mother or father. Therefore, they have a changed/deleted RB1 gene in every somatic cell. A person with an inherited missing or non-functional RB1 gene will develop a retinal tumor if the remaining RB1 gene becomes changed or deleted in a retinal cell. The remaining RB1 gene can become non-functional when exposed to environmental triggers such as chemicals and radiation. In most cases, however, the triggers are unknown. Approximately 90 percent of people who inherit a changed or missing RB1 gene develop retinoblastoma.
People with an inherited form of retinoblastoma are more likely to have a tumor in both eyes (bilateral) and are more likely to have more than one independent tumor (multifocal) in one or both eyes. The average age of onset for the inherited form of retinoblastoma is one year, which is earlier than the sporadic form of retinoblastoma. Although most people with the inherited form of retinoblastoma develop bilateral tumors, approximately 15 percent of people with a tumor in only one eye (unilateral) are affected with an inherited form of retinoblastoma.
A person with an inherited missing or non-functional RB1 gene has a 50 percent chance of passing on this abnormal gene to his or her offspring. The chance that the children will inherit the changed/deleted gene and actually develop retinoblastoma is approximately 45 percent.
Some people with retinoblastoma have inherited a non-functioning or missing RB1 gene from either their mother or father even though their parents have never developed retinoblastoma. It is possible that one parent has a changed or missing RB1 gene in every somatic cell but has not developed retinoblastoma because his or her remaining RB1 gene has remained functional. It is also possible that the parent had developed a retinal tumor that was destroyed by the body. In other cases, one parent has two normal RB1 genes in every somatic cell, but some egg or sperm cells contain a changed or missing RB1 gene. This is called gonadal mosaicism.
Retinoblastoma can also result when both RB1 genes become spontaneously changed or deleted in a retinal cell but the RB1 genes are normal in all the other cells of the body. Approximately 60 percent of people with retinoblastoma have this type of disease, called sporadic retinoblastoma. A person with sporadic retinoblastoma does not have a higher chance of having children with the disease. His or her relatives do not have a higher risk of developing retinoblastoma or having children who develop retinoblastoma. Sporadic retinoblastoma is usually unifocal and has an average age of onset of approximately two years.
Symptoms
The most common symptom of retinoblastoma is leukocoria. Leukocoria results when the pupil reflects a white color rather than the normal black or red color that is seen on a flash photograph. It is often most obvious in flash photographs; since the pupil is exposed to a lot of light and the duration of the exposure is so short, the pupil does not have time to constrict. Children with retinoblastoma can also have problems seeing and this can cause them to appear cross-eyed ( strabismus ). People with retinoblastoma may also experience red, painful, and irritated eyes, inflamed tissue around the eye, enlarged pupils, and possibly different-colored eyes.
Diagnosis
Children who have symptoms of retinoblastoma are usually first evaluated by their pediatrician. The pediatrician will often perform a red reflex test to diagnose or confirm leukocoria. Prior to this test the doctor inserts medicated eye drops into the child's eyes so that the pupils remain dilated and not contract when exposed to bright light. The doctor then examines the eyes with an ophthalmoscope, which shines a bright light into the eyes and allows the doctor to check for leukocoria. Leukocoria can also be diagnosed by taking a flash Polaroid photograph of a patient who has been in a dark room for three to five minutes.
If the pediatrician suspects retinoblastoma on the basis of these evaluations, he or she will most likely refer the patient to an ophthalmologist (eye doctor) who has experience with retinoblastoma. The ophthalmologist will examine the eye using an indirect ophthalmoscope. The opthalmoscope shines a bright light into the eye, which helps the doctor to visualize the retina. This evaluation is usually done under general anesthetic, although some very young or older patients may not require it. Prior to the examination, medicated drops are put into the eyes to dilate the pupils, and anesthetic drops may also be used. A metal clip is used to keep the eyes open during the evaluation. During the examination, a cotton swab or a metal instrument with a flattened tip is used to press on the outer lens of the eye so that a better view of the front areas of the retina can be obtained. Sketches or photographs of the tumor as seen through the ophthalmoscope are taken during the procedure.
An ultrasound evaluation is used to confirm the presence of the tumor and to evaluate its size. Computed axial tomography (CT scan) is used to determine whether the tumor has spread outside of the eye and to the brain. Sometimes magnetic resonance imaging (MRI) is also used to look at the eyes, eye sockets, and the brain to see if the cancer has spread.
In most cases, the cancer has not spread beyond the eye, and other evaluations are unnecessary. If the cancer appears to have spread beyond the eye, then other assessments such as a blood test, spinal tap (lumbar puncture), and/or bone marrow biopsy may be recommended. During a spinal tap, a needle is inserted between the vertebrae of the spinal column and a small sample of the fluid surrounding the spinal cord is obtained. In a bone marrow biopsy, a small amount of tissue (bone marrow) is taken from inside the hip or breast bone for examination.
Genetic testing
Establishing whether someone is affected with an inherited or non-inherited form of retinoblastoma can help to determine whether other family members such as siblings, cousins, and offspring are at increased risk for developing retinoblastoma. It can also sometimes help guide treatment choices, since patients with an inherited form of retinoblastoma may be at increased risk for developing recurrent tumors or other types of cancers, particularly when treated with radiation. It is helpful for the families of a child diagnosed with retinoblastoma to meet with a genetic specialist such as a genetic counselor and/or geneticist. These specialists can help to ascertain the chances that the retinoblastoma is inherited and facilitate genetic testing if desired.
If a patient with unilateral or bilateral retinoblastoma has a relative or relatives with retinoblastoma, it can be assumed that they have an inherited form of retinoblastoma. However, it cannot be assumed that a patient without a family history of the disease has a sporadic form.
Even when there is no family history, most cases of bilateral and trilateral retinoblastoma are inherited, as are most cases of unilateral, multifocal retinoblastoma. However, only 15 percent of unilateral, unifocal retinoblastoma cases are inherited.
The only way to establish whether someone has an inherited form of retinoblastoma is to see if the retinoblastoma gene is changed or deleted in the blood cells obtained from a blood sample. Approximately 5 to 8 percent of individuals with retinoblastoma possess a chromosomal abnormality involving the RB1 gene that can be detected by looking at their chromosomes under the microscope. The chromosomes can be seen by obtaining a blood sample. If this type of chromosomal abnormality is detected in a child, then analysis of the parents' chromosomes should be performed. If one of the parents possesses a chromosomal abnormality, then they are at higher risk for having other offspring with retinoblastoma. Chromosome testing would be recommended for the blood relatives of the parent with the abnormality.
Usually, however, a chromosomal abnormality is not detected in a child with retinoblastoma. In this case, specialized DNA tests that look for small RB1 gene changes need to be performed on the blood cells. DNA testing can be difficult, time consuming, and expensive, since there are many possible RB1 gene changes that can cause the gene to become nonfunctional.
If a sample of tumor is available, then it is recommended that DNA testing be performed on the tumor cells prior to DNA testing of the blood cells. This testing can usually identify the gene changes/deletions in the RB1 genes that caused the tumor to develop. In some cases, RB1 gene changes/deletions are not found in the tumor cells (approximately 20% of RB1 gene changes or deletions are not detectable). In these cases, DNA testing of the blood cells will not be able to ascertain whether someone is affected with an inherited or non-inherited form of retinoblastoma.
If the changes in both RB1 genes are detected in the tumor cell, then these same changes can be looked for in the blood cells. If an RB1 gene is deleted or changed in all of the blood cells tested, the patient can be assumed to have been born with a changed/deleted RB1 gene in all of his or her cells. This person has a 50 percent chance of passing the RB1 gene change/deletion on to his or her children. Most of the time, this change/deletion has been inherited from a parent. Occasionally the gene change/deletion occurred spontaneously in the original cell that was formed when the egg and sperm came together at conception (de novo).
If an RB1 gene change/deletion is found in all of the blood cells tested, both parents should undergo blood testing to check for the same RB1 gene change/deletion. If the RB1 gene change/deletion is identified in one of the parents, it can be assumed that the retinoblastoma was inherited and that siblings have a 50 percent chance of inheriting the altered gene. More distant blood relatives of the parent with the identified RB1 gene change/deletion may also be at risk for developing retinoblastoma. Siblings and other relatives could undergo DNA testing to see if they have inherited the RB1 gene change/deletion.
If the RB1 gene change/deletion is not identified in either parent, then the results can be more difficult to interpret. In this case, there is a 90 to 94 percent chance that the retinoblastoma was not inherited.
In some cases, a person with retinoblastoma will have an RB1 gene change/deletion detected in some of their blood cells and not others. It can be assumed that this person did not inherit the retinoblastoma from either parent. Siblings and other relatives would, therefore, not be at increased risk for developing retinoblastoma. Offspring would be at increased risk since some of the egg or sperm cells could have the changed/deleted RB1 gene. The risks to offspring would probably be less than 50 percent.
In families where there are multiple family members affected with retinoblastoma, blood samples from multiple family members are often analyzed and compared through DNA testing. Ninety-five percent of the time, this type of analysis is able to detect patterns in the DNA that are associated with a changed RB1 gene in that particular family. When a pattern is detected, at-risk relatives can be tested to establish whether they have inherited an RB1 gene change/deletion.
PRENATAL TESTING If chromosome or DNA testing identifies an RB1 gene/deletion in someone's blood cells, then prenatal testing can be performed on this person's offspring. An amniocentesis or chorionic villus sampling can be used to obtain fetal cells which can be analyzed for the RB1 gene change/deletion or chromosomal abnormality.
Treatment
A number of different classification (staging) systems are used to establish the severity of retinoblastoma and aid in choosing an appropriate treatment plan. The most widely used staging system is the Reese-Ellsworth system. This system is used to classify intraocular tumors and predict which tumors are favorable enough that sight can be maintained. The Reese-Ellsworth classification system is divided into several groups:
- Group I (very favorable for maintenance of sight): small solitary or multiple tumors, less than 6.4 mm in size (1 inch equals 25.4 mm), located at or below the equator of the eye.
- Group II (favorable for maintenance of sight): solitary or multiple tumors, 6.4 to 16 mm in size, located at or behind the equator of the eye.
- Group III (possible for maintenance of sight): any tumor located in front of the equator of the eye, or a solitary tumor larger than 16 mm in size and located behind the equator of the eye.
- Group IV (unfavorable for maintenance of sight): multiple tumors, some larger than 16 mm in size, or any tumor extending in front of the outer rim of the retina (ora serrata).
- Group V (very unfavorable for maintenance of sight): large tumors involving more than half of the retina, or vitreous seeding, in which small pieces of tumor are broken off and floating around the inside of the eye.
When choosing a treatment plan, the first criterion is to determine whether the cancer is localized within the eye (intralocular) or has spread to other parts of the body (extralocular). An intraocular retinoblastoma may only involve the retina or could involve other parts of the eye. An extraocular retinoblastoma could involve only the tissues around the eye or could result from the spread of cancer to the brain or other parts of the body.
It is also important to establish whether the cancer is unilateral (one eye) or bilateral (both eyes), multifocal or unifocal. In order for the tumors to be considered multifocal, they must have arisen independently and not as the result of the spread of cancer cells. It is also important to check for trilateral retinoblastoma.
The treatment chosen depends on the size and number of tumors, whether the cancer is unilateral or bilateral, and whether the cancer has spread to other parts of the body. The goal of treatment is to cure the cancer and prevent as much loss of vision as possible.
TREATMENT OF INTRAOCULAR TUMORS Surgical removal of the affected eye (enucleation) is performed when the tumor(s) are so large and extensive that preservation of sight is not possible. This surgery is performed under general anesthetic and usually takes less than an hour. Most children who have undergone this surgery can leave the hospital on the same day. A temporary ball is placed in the eye socket after the surgery. Approximately three weeks after the operation, a plastic artificial eye (prosthesis) that looks like the normal eye is inserted into the eye socket.
Radiation therapy is often used for treatment of large tumors when preservation of sight is possible. External beam radiation therapy involves focusing a beam of radiation on the eye. If the tumor has not spread extensively, the radiation beam can be focused on the cancerous retinal cells. If the cancer is extensive, radiation treatment of the entire eye may be necessary. External beam radiation is performed on an outpatient basis and usually occurs over a period of three to four weeks.
Some children may need sedatives prior to the treatment. This type of therapy can result in a temporary loss of a patch of hair on the back of the head and a small area of "sun-burned" skin. Long-term side effects of radiation treatment can include cataracts, vision problems, bleeding from the retina, and decreased growth of the bones on the side of the head. People with an inherited form of retinoblastoma have an increased risk of developing other cancers as a result of this therapy. Some consideration should, therefore, be given to alternative treatment therapies for those with an inherited form of retinoblastoma.
Photocoagulation therapy is often used in conjunction with radiation therapy but may be used alone to treat small tumors that are located on the back of the eye. Photocoagulation involves using a laser to destroy the cancer cells. This type of treatment is performed under local or general anesthesia and is usually not associated with post-procedural pain .
Thermotherapy is also often used in conjunction with radiation therapy or drug therapy ( chemotherapy ). Thermotherapy involves the use of heat to help shrink tumor cells. The heat is either used on the whole eye or localized to the tumor area. It is performed under local or general anesthesia and is usually not painful.
Cryotherapy is a treatment often used in conjunction with radiation therapy but can also be used alone on small tumors located on the front part of the retina. Cryotherapy involves the use of intense cold to destroy cancer cells and can result in harmless, temporary swelling of the external eye and eyelids that can last for up to five days. Eye drops or ointment are sometimes provided to reduce the swelling.
Brachytherapy involves the application of radioactive material to the outer surface of the eye at the base of the tumor. It is generally used for tumors of medium size. A patient undergoing this type of procedure is usually hospitalized for three to seven days. During that time, he or she undergoes one surgery to attach the radioactive material and one surgery to remove it. Eye drops are often administered for three to four weeks following the operation to prevent inflammation and infection. The long-term side effects of this treatment can include cataracts and damage to the retina, which can lead to impaired vision.
Intravenous treatment with one or more drugs (chemotherapy) is often used for treatment of both large and small tumors. Chemotherapy is sometimes used to shrink tumors prior to other treatments such as radiation therapy or brachytherapy. Occasionally, it is also used alone to treat very small tumors.
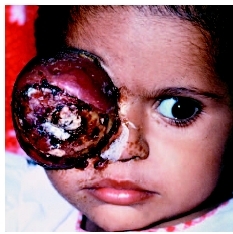
TREATMENT OF INTRAOCULAR AND UNILATERAL RETINOBLASTOMA Often, by the time that unilateral retinoblastoma is diagnosed, the tumor is so large that useful vision cannot be preserved. In these cases removal of the eye (enucleation) is the treatment of choice. Other therapies are unnecessary if enucleation is used to treat intraocular unilateral retinoblastoma. If the tumor is small enough, other therapies such as external beam radiation therapy, photocoagulation, cryotherapy, thermotherapy, chemotherapy, and brachytherapy may be considered.
TREATMENT OF INTRAOCULAR AND BILATERAL RETINOBLASTOMA If vision can be preserved in both eyes, radiation therapy of both eyes may be recommended. Smaller, more localized tumors can sometimes be treated by local therapies such as cryotherapy, photocoagulation therapy, thermotherapy or brachytherapy. Some centers may use chemotherapy in place of radiation therapy when the tumors are too large to be treated by local therapies or are found over the optic nerve of the eye. As of the early 2000s, many centers are moving away from radiation treatment and toward chemotherapy because it is less likely to induce future tumors. Enucleation is performed on the more severely affected eye if sight cannot be preserved in both.
EXTRAOCULAR RETINOBLASTOMA There is no proven effective therapy for the treatment of extraocular retinoblastomas. Commonly, radiation treatment of the eyes and chemotherapy is provided.
Alternative treatment
As of 2004 there are no alternative or complementary therapies specific to the treatment of retinoblastoma. Since most people diagnosed with retinoblastoma are small children, most drug-based alternative therapies designed to treat general cancer would not be recommended. Many specialists would, however, stress the importance of establishing a well-balanced diet, including certain fruits, vegetables, and vitamin supplements, to ensure that the body is strengthened in its fight against cancer. Some advocate the use of visualization strategies, in which patients are encouraged to visualize the immune cells of their body attacking and destroying the cancer cells.
Prognosis
Individuals with intraocular retinoblastoma who do not have trilateral retinoblastoma usually have a good survival rate with a 90 percent chance of disease-free survival for five years. Those with extraocular retinoblastoma have less than a 10 percent chance of disease-free survival for the same amount of time. Trilateral retinoblastoma generally has a very poor prognosis. Patients with trilateral retinoblastoma who receive treatment have an average survival rate of approximately eight months, while those who remain untreated have an average survival rate of approximately one month. Patients with trilateral retinoblastoma who are asymptomatic at the time of diagnosis may have a better prognosis then those who experience symptoms.
Patients with an inherited form of unilateral retinoblastoma have a 70 percent chance of developing retinoblastoma in the other eye. Retinoblastoma reoccurs in the other eye in approximately 5 percent of people with a non-inherited form of retinoblastoma, so it is advisable for even these patients to be closely monitored. People with an inherited form of retinoblastoma who have not undergone radiation treatment have approximately a 26 percent chance of developing cancer in another part of the body within 50 years of the initial diagnosis. Those with an inherited form who have undergone radiation treatment have a 58 percent chance of developing a secondary cancer within 50 years after the initial diagnosis. Most of the secondary cancers are skin cancers, bone tumors (osteosarcomas), and soft-tissue sarcomas . Soft-tissue sarcomas are malignant tumors of the muscle, nerves, joints, blood vessels, deep skin tissues, or fat.
Prevention
Although retinoblastoma cannot be prevented, appropriate screening and surveillance should be applied to
KEY TERMS
Amniocentesis —A procedure performed at 16–18 weeks of pregnancy in which a needle is inserted through a woman's abdomen into her uterus to draw out a small sample of the amniotic fluid from around the baby for analysis. Either the fluid itself or cells from the fluid can be used for a variety of tests to obtain information about genetic disorders and other medical conditions in the fetus.
Benign tumor —An abnormal proliferation of cells that does not spread to other parts of the body.
Bilateral —Occurring on two sides. For example, a patient with bilateral retinoblastoma has this retinal tumor in both eyes.
Brachytherapy —A method of treating cancers, such as prostate cancer, involving the implantation near the tumor of radioactive seeds.
Chorionic villus sampling —A procedure used for prenatal diagnosis at 10–12 weeks gestation. Under ultrasound guidance a needle is inserted either through the mother's vagina or abdominal wall and a sample of the chorionic membrane. These cells are then tested for chromosome abnormalities or other genetic diseases.
Chromosome —A microscopic thread-like structure found within each cell of the human body and consisting of a complex of proteins and DNA. Humans have 46 chromosomes arranged into 23 pairs. Chromosomes contain the genetic information necessary to direct the development and functioning of all cells and systems in the body. They pass on hereditary traits from parents to child (like eye color) and determine whether the child will be male or female.
Cryotherapy —The use of a very low-temperature probe to freeze and thereby destroy tissue. Cryotherapy is used in the treatment skin lesions, Parkinson's disease, some cancers, retinal detachment, and cataracts. Also called cryosurgery.
DNA —Deoxyribonucleic acid; the genetic material in cells that holds the inherited instructions for growth, development, and cellular functioning.
DNA testing —Analysis of DNA (the genetic component of cells) in order to determine changes in genes that may indicate a specific disorder.
Enucleation —Surgical removal of the eyeball.
Equator —Imaginary line encircling the eyeball and dividing the eye into a front and back half.
Extraocular retinoblastoma —Cancer that has spread from the eye to other parts of the body.
Gene —A building block of inheritance, which contains the instructions for the production of a particular protein, and is made up of a molecular sequence found on a section of DNA. Each gene is found on a precise location on a chromosome.
Intraocular retinoblastoma —Cancer of the retina that is limited to the eye and has not spread to other parts of the body.
Malignant tumor —An abnormal proliferation of cells that can spread to other sites.
Multifocal —Having many focal points. When referring to a disease, it means that damage caused by the disease occurs at multiple sites. When referring to a cancer, it means that more than one tumor is present.
Oncologist —A physician specializing in the diagnosis and treatment of cancer
Ophthalmologist —A physician who specializes in the anatomy and physiology of the eyes and in the diagnosis and treatment of eye diseases and disorders.
Optic nerve —A bundle of nerve fibers that carries visual messages from the retina in the form of electrical signals to the brain.
Photocoagulation —A type of cancer treatment in which cancer cells are destroyed by an intense beam of laser light.
Prenatal testing —Testing for a disease, such as a genetic condition, in an unborn baby.
Protein —An important building blocks of the body, a protein is a large, complex organic molecule composed of amino acids. It is involved in the formation of body structures and in controlling the basic functions of the human body.
Retina —The inner, light-sensitive layer of the eye containing rods and cones. The retina transforms the image it receives into electrical signals that are sent to the brain via the optic nerve.
Somatic cells —All the cells of the body with the exception of the egg and sperm cells.
Tumor —A growth of tissue resulting from the uncontrolled proliferation of cells.
Tumor-suppressor gene —A gene involved in controlling normal cell growth and preventing cancer.
Unifocal —Only one tumor present in one eye.
Unilateral —Refers to one side of the body or only one organ in a pair.
Vitreous —The transparent gel that fills the back part of the eye.
Vitreous seeding —Small pieces of tumor have broken off and are floating around the vitreous.
all at-risk individuals to ensure that the tumor(s) are diagnosed at an early stage. The earlier the diagnosis, the more likely that an eye can be salvaged and vision maintained.
Screening of people diagnosed with retinoblastoma
Children who have been diagnosed with retinoblastoma should receive periodic dilated retinal examinations until the age of five. Young children will need to undergo these evaluations under anesthetic. After five years of age, periodic eye examinations are recommended. It may be advisable for patients with bilateral retinoblastoma or an inherited form of retinoblastoma to undergo periodic screening for the brain tumors found in trilateral retinoblastoma. There are no specific screening protocols designed to detect non-ocular tumors. All lumps and complaints of bone pain, however, should be thoroughly evaluated.
Screening of relatives
When a child is diagnosed with retinoblastoma, it is recommended that parents and siblings receive a dilated retinal examination by an ophthalmologist who is experienced in the diagnosis and treatment of the disease. It is also recommended that siblings continue to undergo periodic retinal examinations under anesthetic until they are three years of age. For children three to seven years of age, periodic eye examinations are recommended. The retinal examinations can be avoided if DNA testing indicates that the patient has a non-inherited form of retinoblastoma or if the sibling has not inherited the RB1 gene change/deletion. Any relatives who are found through DNA testing to have inherited an RB1 gene change/deletion should undergo the same surveillance procedures as siblings.
The children of someone diagnosed with retinoblastoma should also undergo periodic retinal examinations under anesthetic. Retinal surveillance should be performed unless DNA testing proves that their child does not possess the RB1 gene change/deletion. If desired, prenatal detection of tumors using ultrasound may also be performed. During the ultrasound procedure, a hand-held instrument is placed on the maternal abdomen or inserted vaginally. The ultrasound produces sound waves that are reflected back from the body structures of the fetus, producing a picture that can be seen on a video screen. If a tumor is detected through this evaluation, the affected baby may be delivered a couple of weeks earlier. This can allow for earlier intervention and treatment.
Parental concerns
Careful attention to a child's diet can be very helpful for patients with cancer. This can be difficult when the cancer and/or the treatments are affecting the appetite, however. Whole foods, including grains, beans, fresh fruits and vegetables, and high quality fats, should be emphasized in the diet, while processed foods should be avoided. Increased consumption of fish, especially cold-water fish like salmon, mackerel, halibut, and tuna, provides a good source of omega-3 fatty acids. Nutritional supplements can build strength and help maintain it during and following chemotherapy, radiation, or surgery.
Guided imagery and relaxation techniques can be helpful for children undergoing difficult treatments. Support groups for the child and the family can be very helpful and can provide an important emotional outlet for the child, the parents, and the siblings.
Resources
BOOKS
Elner, Victor M. et al. "Eye, Orbit, and Adnexal Structures." In Clinical Oncology. Edited by Martin D. Abeloff. London: Churchill Livingstone, 2000.
Herzog, Cynthia E. "Retinoblastoma." In Nelson Textbook of Pediatrics. Edited by Richard E. Behrman et al. Philadelphia: Saunders, 2004.
PERIODICALS
Castillo, B. V. "Pediatric tumors of the eye and orbit." Pediatric Clinics of North America 50 (February 2003): 149–72.
Pakakasama, S. "Genetic predisposition and screening in pediatric cancer." Pediatric Clinics of North America 49 (December 2002): 1393–413.
Pratt, C. B. "Successful treatment of intraocular retinoblastoma with chemotherapy." Journal of Pediatrics 140 (May 2002): 635.
ORGANIZATIONS
Institute for Families with Blind Children. PO Box 54700, Mail Stop 111, Los Angeles, CA 90054–0700. Web site: http://www.instituteforfamilies.org.
Retinoblastoma International. 4650 Sunset Blvd., Mail Stop #88, Los Angeles, CA 90027. Web site: http://www.retinoblastoma.net.
WEB SITES
"Retinoblastoma." CancerNet. Available online at http://cancernet.nci.nih.gov/cancertopics/types/retinoblastoma (accessed December 30, 2004).
Lisa Andres, MS, CGC Rosalyn Carson-DeWitt, MD
i have many picture of retinbalstoma if u're interested