Ehlers-Danlos syndrome
Definition
The Ehlers-Danlos syndrome (EDS) refers to a group of inherited disorders that affect collagen structure and function. Genetic abnormalities in the manufacturing of collagen within the body affect connective tissues, causing them to be abnormally weak. Ehlers-Danlos syndrome is also referred to as inherited connective tissue disorder.
Description
Collagen is a strong, fibrous protein that lends strength and elasticity to connective tissues such as the skin, tendons, organ walls, cartilage, and blood vessels. Each of these connective tissues requires collagen tailored to meet its specific purposes. The many roles of collagen are reflected in the number of genes dedicated to its production. There are at least 28 genes in humans that encode at least 19 different types of collagen. Mutations in these genes can affect basic construction as well as the fine-tuned processing of the collagen.
EDS is a group of inherited connective tissue disorders that usually affects the skin, ligaments, joints, and blood vessels. Classification of EDS types was revised in 1997. The new classification involves categorizing the different forms of EDS into six major sub-types, including classical, hypermobility, vascular, kyphoscoliosis, arthrochalasia, and dermatosparaxis, and a collection of rare or poorly defined varieties. This new classification is simpler than the previous classification system and is primarily based on descriptions of the actual symptoms.
Classical type
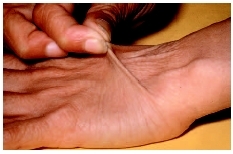
Under the old classification system, EDS classical type was divided into two separate types: type I and type II. The major symptoms associated with EDS classical type involve the skin and joints. The skin has a smooth, velvety texture, and bruises easily. Affected individuals typically have extensive scarring, particularly at the knees, elbows, forehead, and chin. The joints are hyperextensible, giving a tendency toward dislocation of the hip, shoulder, elbow, knee, or clavicle. Due to decreased muscle tone, affected infants may experience a delay in reaching motor milestones. Children may have a tendency to develop hernias or other organ shifts within the abdomen. Sprains and partial or complete joint dislocations are also common. Symptoms can range from mild to severe. EDS classical type is inherited in an autosomal dominant manner.
There are three major clinical diagnostic criteria for EDS classical type: skin hyperextensibility, unusually wide scars, and joint hypermobility. There is no definitive test for the diagnosis of classical EDS. Both DNA and biochemical studies have been used to help identify affected individuals. In some cases, skin biopsy has been found useful in confirming a diagnosis. Unfortunately, these tests are not sensitive enough to identify all individuals with classical EDS. If there are multiple affected individuals in a family , it may be possible to perform prenatal diagnosis using a DNA information technique known as a linkage study.
Hypermobility type
Excessively loose joints are the hallmark of this EDS type, formerly known as EDS type III. Both large joints, such as the elbows and knees, and small joints, such as toes and fingers, are affected. Partial and total joint dislocations are common, particularly involving the jaw, knee, and shoulder. Many individuals experience chronic limb and joint pain , although x rays of these joints appear normal. The skin may also bruise easily. Osteoarthritis is a common occurrence in adults. EDS hypermobility type is inherited in an autosomal dominant manner.
There are two major clinical diagnostic criteria for EDS hypermobility type. These include skin involvement (either hyperextensible skin or smooth and velvety skin) and generalized joint hypermobility. There is no test for this form of EDS.
Vascular type
Formerly called EDS type IV, EDS vascular type is the most severe form. The connective tissue in the intestines, arteries, uterus, and other hollow organs may be unusually weak, leading to organ or blood vessel rupture. Such ruptures most likely occur between ages 20 and 40, although they can occur any time, and may be life-threatening.
There is a classic facial appearance associated with EDS vascular type. Affected individuals tend to have large eyes, a thin pinched nose, thin lips, and a slim body. The skin is thin and translucent, with veins dramatically visible, particularly across the chest.
The large joints have normal stability, but small joints in the hands and feet are loose, showing hyperextensibility. The skin bruises easily. Other complications may include collapsed lungs, premature aging of the skin on the hands and feet, and ruptured arteries and veins. After surgery, there may be poor wound healing, a complication that tends to be frequent and severe. Pregnancy also carries the risk of complications. During and after pregnancy, there is an increased risk of the uterus rupturing and of arterial bleeding. Due to the severe complications associated with EDS type IV, death usually occurs before the fifth decade. A study of 419 individuals with EDS vascular type, completed in 2000, found that the median survival rate was 48 years, with a range of six to 73 years. EDS vascular type is inherited in an autosomal dominant manner.
There are four major clinical diagnostic criteria for EDS vascular type. These include thin translucent skin, arterial/intestinal/uterine fragility or rupture, extensive bruising, and characteristic facial appearance. EDS vascular type is caused by a change in the gene COL3A1, which codes for one of the collagen chains used to build Collagen type III. Laboratory testing is available for this form of EDS. A skin biopsy may be used to demonstrate the structurally abnormal collagen. This type of biochemical test identifies more than 95 percent of individuals with EDS vascular type. Laboratory testing is recommended for individuals with two or more of the major criteria.
DNA analysis may also be used to identify the change within the COL3A1 gene. This information may be helpful for genetic counseling purposes. Prenatal testing is available for pregnancies in which an affected parent has been identified and the DNA mutation is known or the biochemical defect has been demonstrated.
Kyphoscoliosis type
The major symptoms of kyphoscoliosis type, formerly called EDS type VI, are general joint looseness. At birth, the muscle tone is poor, and motor skill development is subsequently delayed. Also, infants with this type of EDS have an abnormal curvature of the spine ( scoliosis ). The scoliosis becomes progressively worse with age; affected individuals are usually unable to walk by age 20. The eyes and skin are fragile and easily damaged, and blood vessel involvement is a possibility. The bones may also be affected as demonstrated by a decrease in bone mass. Kyphoscoliosis type is inherited in an autosomal recessive manner.
There are four major clinical diagnostic criteria for EDS kyphoscoliosis type. These include generally loose joints, low muscle tone at birth, scoliosis at birth (which worsens with age), and a fragility of the eyes, which may give the white area of the eye a blue tint or cause the eye to rupture. This form of EDS is caused by a change in the PLOD gene on chromosome 1, which encodes the enzyme lysyl hydroxylase. A laboratory test is available in which urinary hydroxylysyl pryridinoline is measured. This test, performed on urine, is extremely sensitive and specific for EDS kyphoscolios type. Laboratory testing is recommended for infants with three or more of the major diagnostic criteria.
Prenatal testing is available if a pregnancy is known to be at risk and an identified affected family member has had positive laboratory testing. An amniocentesis may be performed in which fetal cells are removed from the amniotic fluid and enzyme activity is measured.
Arthrochalasia type
Dislocation of the hip joint typically accompanies arthrochalasia type EDS, formerly called EDS type VIIB. Other joints are also unusually loose, leading to recurrent partial and total dislocations. The skin has a high degree of stretchability and bruises easily. Individuals with this type of EDS may experience mildly diminished bone mass, scoliosis, and poor muscle tone. Arthrochalasia type is inherited in an autosomal dominant manner.
There are two major clinical diagnostic criteria for EDS arthrochalasia type. These include severe generalized joint hypermobility and bilateral hip dislocation present at birth. This form of EDS is caused by a change in either of two components of Collagen type I, called proa1(I) type A and proa2(I) type B. A skin biopsy may be performed to demonstrate an abnormality in either component. Direct DNA testing is also available.
Dermatosparaxis type
Individuals with this type of EDS, once called type VIIC, have extremely fragile skin that bruises easily but does not scar excessively. The skin is soft and may sag, leading to an aged appearance even in young adults. Individuals may also have hernias. Dermatosparaxis type is inherited in an autosomal recessive manner.
There are two major clinical diagnostic criteria for EDS dematosparaxis type. These include severe skin fragility and sagging or aged-appearing skin. This form of EDS is caused by a change in the enzyme called procollagen I N-terminal peptidase. A skin biopsy may be performed for a definitive diagnosis of dermatosparaxis type.
Other types
There are several other forms of EDS that have not been as clearly defined as the aforementioned types. Symptoms of EDS within this category may include soft, mildly stretchable skin, shortened bones, chronic diarrhea , joint hypermobility and dislocation, bladder rupture, or poor wound healing. Inheritance patterns within this group include X-linked recessive, autosomal dominant, and autosomal recessive.
Demographics
EDS was originally described by Dr. Van Meekeren in 1682. Dr. Ehlers and Dr. Danlos further characterized the disease in 1901 and 1908, respectively. According to the Ehlers-Danlos National Foundation, one in 5,000 to one in 10,000 people are affected by some form of EDS.
Causes and symptoms
There are numerous types of EDS, all caused by changes in one of several genes. The manner in which EDS is inherited depends on the specific gene involved. There are three patterns of inheritance for EDS: autosomal dominant, autosomal recessive, and X-linked (extremely rare).
Chromosomes are made up of hundreds of small units known as genes, which contain the genetic material necessary for an individual to develop and function. Humans have 46 chromosomes, which are matched into 23 pairs. Because chromosomes are inherited in pairs, each individual receives two copies of each chromosome and likewise two copies of each gene.
Changes or mutations in genes can cause genetic diseases in several different ways, many of which are represented within the spectrum of EDS. In autosomal dominant EDS, only one copy of a specific gene must be changed for a person to have EDS. In autosomal recessive EDS, both copies of a specific gene must be changed for a person to have EDS. If only one copy of an autosomal recessive EDS gene is changed, the person is referred to as a carrier, meaning he or she does not have any signs or symptoms of the disease itself, but carries the possibility of passing the disorder to a future child. In X-linked EDS, a specific gene on the X chromosome must be changed. However, this affects males and females differently because males and females have a different number of X chromosomes.
The few X-linked forms of EDS fall under the category of X-linked recessive. As with autosomal recessive, this implies that both copies of a specific gene must be changed for a person to be affected. However, because males only have one X-chromosome, they are affected if an X-linked recessive EDS gene is changed on their single X-chromosome. That is, they are affected even though they have only one changed copy. On the other hand, that same gene must be changed on both of the X-chromosomes in a female for her to be affected.
Although there is much information regarding the changes in genes that cause EDS and their various inheritance patterns, the exact gene mutation for all types of EDS is not known.
When to call the doctor
The doctor should be called if a child has symptoms of Ehlers-Danlos syndrome. Medical advice should also be sought if a person has a family history of Ehlers-Danlos syndrome and is planning to conceive a child.
Diagnosis
Clinical symptoms such as extreme joint looseness and unusual skin qualities, along with family history, can lead to a diagnosis of EDS. Specific tests, such as skin biopsies, are available for diagnosis of certain types of EDS, including vascular, arthrochalasia, and dermatosparaxis types. A skin biopsy involves removing a small sample of skin and examining its microscopic structure. A urine test is available for the kyphoscoliosis type.
Treatment
Medical therapy relies on managing symptoms and trying to prevent further complications. There is no cure for EDS.
Braces may be prescribed to stabilize joints, although surgery is sometimes necessary to repair joint damage caused by repeated dislocations. Physical therapy teaches individuals how to strengthen muscles around joints and may help to prevent or limit damage. Elective surgery is discouraged due to the high possibility of complications.
There are anecdotal reports that large daily doses (0.04–0.14 oz, or 1–4 g) of vitamin C may help decrease bruising and aid in wound healing. Constitutional homeopathic treatment may be helpful in maintaining optimal health in persons with a diagnosis of EDS. Before beginning these types of therapies, an individual with EDS should discuss them with his or her doctor. Therapy that does not require medical consultation
Prognosis
The outlook for individuals with EDS depends on the type of EDS with which they have been diagnosed. Symptoms vary in severity, even within one sub-type. Some individuals have negligible symptoms, while others are severely restricted in their daily life. Extreme joint instability and scoliosis may limit a person's mobility. Most individuals will have a normal lifespan. However, those with blood vessel involvement, particularly persons with EDS vascular type, have an increased risk of fatal complications.
EDS is a lifelong condition. Affected individuals may face social obstacles related to their disease on a daily basis. Some individuals with EDS have reported living with fears of significant and painful skin ruptures, becoming pregnant (especially those with EDS vascular type), experiencing worsening of their condition, becoming unemployed due to physical and emotional burdens, and undergoing social stigmatization in general.
Some people with EDS are not diagnosed until well into adulthood and, in the case of EDS vascular type, occasionally not until after death due to complications of the disorder. The diagnosis may be devastating to the individual and, in many cases, to other family members when they learn they are at risk for being affected.
Although children with EDS face significant challenges, it is important to remember that each child is unique with his or her own distinguished qualities and potential. Persons with EDS go on to have families and careers, and to be respected citizens, surmounting the challenges of their disease.
Prevention
If a couple has had a child diagnosed with EDS, the chance that they will have another child with the same disorder depends on the form of EDS the child has and if either parent is affected by the same disease.
In classical autosomal dominant EDS, the risk for recurrence in the parents' other children is one in four.
X-linked recessive EDS is accompanied by a slightly more complicated pattern of inheritance. If a father with an X-linked recessive form of EDS passes a copy of his X chromosome to his children, the sons will be unaffected and the daughters will be carriers. If a mother is a carrier for an X-linked recessive form of EDS, she may have affected or unaffected sons, or carrier or unaffected daughters, depending on the second sex chromosome inherited from the father.
Prenatal diagnosis is available for specific forms of EDS, including kyphosocliosis type and vascular type. However, prenatal testing is only a possibility in these types if the underlying defect has been found in another family member.
Parental concerns
Constant bruises, skin wounds, and trips to the hospital take their toll on affected children and their parents. Prior to diagnosis, parents of children with EDS have found themselves under suspicion of child abuse .
KEY TERMS
Arthrochalasia —Excessive loosness of the joints.
Blood vessels —General term for arteries, veins, and capillaries that transport blood throughout the body.
Cartilage —A tough, elastic connective tissue found in the joints, outer ear, nose, larynx, and other parts of the body.
Collagen —The main supportive protein of cartilage, connective tissue, tendon, skin, and bone.
Connective tissue —A group of tissues responsible for support throughout the body; includes cartilage, bone, fat, tissue underlying skin, and tissues that support organs, blood vessels, and nerves throughout the body.
Dermatosparaxis —Skin fragility caused by abnormal collagen.
Hernia —A rupture in the wall of a body cavity, through which an organ may protrude.
Homeopathy —A holistic system of treatment developed in the eighteenth century. It is based on the idea that substances that produce symptoms of sickness in healthy people will have a curative effect when given in very dilute quantities to sick people who exhibit those same symptoms. Homeopathic remedies are believed to stimulate the body's own healing processes.
Hyperextensibility —The ability to extend a joint beyond the normal range.
Hypermobility —Unusual flexibility of the joints, allowing them to be bent or moved beyond their normal range of motion.
Joint dislocation —The displacement of a bone from its socket or normal position.
Kyphoscoliosis —Abnormal front-to-back and side-to-side curvature of the spine.
Ligament —A type of tough, fibrous tissue that connects bones or cartilage and provides support and strength to joints.
Osteoarthritis —A noninflammatory type of arthritis, usually occurring in older people, characterized by degeneration of cartilage, enlargement of the margins of the bones, and changes in the membranes in the joints. Also called degenerative arthritis.
Scoliosis —An abnormal, side-to-side curvature of the spine.
Tendon —A tough cord of dense white fibrous connective tissue that connects a muscle with some other part, especially a bone, and transmits the force which the muscle exerts.
Uterus —The female reproductive organ that contains and nourishes a fetus from implantation until birth. Also called the womb.
Vascular —Pertaining to blood vessels.
Management of all types of EDS may include genetic counseling to help the affected individual and his or her family understand the disorder and its impact on other family members and future children.
Resources
BOOKS
Icon Health Publications. Ehlers-Danlos Syndrome: A Medical Dictionary, Bibliography, and Annotated Research Guide to the Internet. San Diego, CA: Icon Health Publications, 2004.
——. The Official Patient's Sourcebook on Ehlers-Danlos Syndrome: A Revised and Updated Directory for the Internet Age. San Diego, CA: Icon Health Publications, 2002.
Massoud, Lindalee. Ehlers-Danlos Syndrome: Medical and Practical Information. Flint, MI: SignQuest Publishers, 1997.
PM Medical Health News. 21st Century Complete Guide to Ehlers-Danlos Syndrome (EDS), Connective Tissue Disorders, Hypermobility, Authoritative Government Documents, Clinical References, and Practical Information for Patients and Physicians. Washington, DC: Progressive Management, 2004.
PERIODICALS
"Living a Restricted Life with Ehlers-Danlos Syndrome." International Journal of Nursing Studies 37 (2000): 111–118.
ORGANIZATIONS
Ehlers-Danlos Support Group - UK. P.O. Box 335, Farnham, Surrey, GU10 1XJ. UK. +44 1252 690 940. Web site: http://www.ehlers-danlos.org.
Ehlers-Danlos National Foundation. 6399 Wilshire Blvd., Ste. 203, Los Angeles, CA 90048 (323) 651-3038. Fax: (323) 651-1366. Web site: http://www.ednf.org.
WEB SITES
GeneClinics. Available online at: http://www.geneclinics.org.
Judith Sims Java O. Solis, M.S.
Comment about this article, ask questions, or add new information about this topic: