Hemophilia
Definition
Hemophilia is a coagulation disorder arising from a genetic defect of the X chromosome; the defect can either be inherited or result from spontaneous gene mutation. In each type of hemophilia (hemophilias A, B, and C), a critical coagulation protein is missing, causing individuals to bleed for long periods of time before clotting occurs. Depending on the degree of the disorder in the affected individual, uncontrolled bleeding may occur spontaneously with no known initiating event, or occur after specific events such as surgery, dental procedures, immunizations, or injury.
Description
The body's normal mechanism for blood clotting is a complex series of events (coagulation cascade) involving interaction between the injured blood vessel, blood cells called platelets, 13 specific coagulation factors (designated by Roman numerals I through XIII), and other substances that circulate in the blood.
When blood vessels are injured in a way that causes bleeding, platelets collect over the injured area, forming a temporary plug to prevent further bleeding. This temporary plug, however, is too disorganized to serve as a long-term solution, so a series of chemical events occurs that results in the formation of a more reliable plug. The final plug or clot involves tightly woven fibers of a material called fibrin. The production of fibrin requires the interaction of a series of proteins, clotting factors I through XIII, in a process called amplification to rapidly produce the proper-sized fibrin clot from the small number of molecules initially activated by the injury. In the complex coagulation process, the absence or inactivity of just one clotting factor can greatly increase bleeding time. In hemophilia, certain clotting factors are either decreased in quantity, absent altogether, or improperly formed, preventing the formation of a clot and resulting in uncontrolled bleeding.
Hemophilia A is the most common type of coagulation disorder and involves decreased activity of factor VIII. There are three levels of factor VIII deficiency: severe, moderate, and mild. This classification is based on the percentage of normal factor VIII activity present:
- Individuals with less than 1 percent of normal factor VIII activity level have severe hemophilia. Half of all people with hemophilia A fall into this category. Such individuals frequently experience spontaneous musculoskeletal bleeding into their joints, skin, and muscles. Surgery or trauma can result in life-threatening hemorrhage and must be carefully managed.
- Individuals with 1–5 percent of normal factor VIII activity level have moderate hemophilia and are at risk for heavy bleeding after seemingly minor traumatic injuries.
- Individuals with 5–40 percent of normal factor VIII activity level have mild hemophilia and must prepare carefully for any surgery or dental procedures.
In hemophilia B, or Christmas disease, the deficient clotting factor is factor IX, but the symptoms are very similar to those of hemophilia A. Factor IX is produced in the liver and is dependent on interaction with vitamin K in order to function properly. A deficiency in vitamin K can affect the clotting factor's performance as well as a deficiency in the factor itself.
Hemophilia C is rare and much milder than hemophilia A or B. It involves reduced activity of factor XI and is characterized by mild bleeding such as nosebleeds (epistaxis) or prolonged menstrual bleeding, or mild bleeding after tonsillectomies or dental extractions.
Demographics
Hemophilia A affects between one in 5,000 to one in 10,000 males in most populations. Hemophilia B occurs in one in 40,000 to 50,000. The prevalence of hemophilia is estimated to be 13.4 cases per 100,000 U.S. males (10.5 hemophilia A and 2.9 hemophilia B). By race/ethnicity, the prevalence is 13.2 cases in 100,000 among white males, 11.0 among African-American males, and 11.5 among Hispanic males. Hemophilia C occurs primarily among individuals of Jewish descent.
Causes and symptoms
Hemophilia A and B are both caused by a genetic defect present on the X chromosome. (Hemophilia C is inherited in a different fashion.) About 70 percent of all people with hemophilia A or B inherited the disease. The other 30 percent develop from a spontaneous genetic mutation.
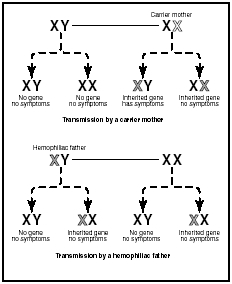
Both factors VIII and IX are produced by a genetic defect of the X chromosome, so hemophilia A and B are both sex-linked diseases passed on from a female to male offspring. (All humans have two chromosomes determining their gender: females have XX, males have XY. Because the trait is carried only on the X chromosome, it is called sex-linked.) Because a female child always receives two X chromosomes, she will nearly always receive at least one normal X chromosome. Therefore, even if she receives one flawed X chromosome, she will still be capable of producing a sufficient quantity of factors VIII and IX to avoid the symptoms of hemophilia. Such a person who has one flawed chromosome but does not actually suffer from the disease is called a carrier. She carries the flaw that causes hemophilia and can pass it on to her offspring. If, however, she has a son who receives her flawed X chromosome, he will be unable to produce the right quantity of factors VIII or IX, and he will suffer some degree of hemophilia. (Males inherit one X and one Y chromosome and, therefore, have only one X chromosome.)
In rare cases, a hemophiliac father and a carrier mother can pass on the right combination of parental chromosomes to result in a hemophiliac female child. However, the vast majority of people with either hemophilia A or B are male.
About 30 percent of all people with hemophilia A or B are the first member of their family to ever have the disease. These individuals have had the unfortunate occurrence of a spontaneous mutation, meaning that in their early development, some random genetic accident affected their X chromosome, resulting in the defect that causes hemophilia A or B. Once such a spontaneous genetic mutation takes place, offspring of the affected person can inherit the newly created, flawed chromosome.
In the case of severe hemophilia, the first bleeding event usually occurs prior to 18 months of age. In some babies, hemophilia is suspected immediately when a routine circumcision (removal of the foreskin of the penis) results in unusually heavy bleeding. Toddlers are at particular risk because they fall frequently and may bleed into the soft tissue of their arms and legs. These small bleeds result in bruising and noticeable lumps but do not usually require treatment. As a child becomes more active, bleeding may occur into the muscles, a much more painful and debilitating situation. These muscle bleeds result in pain and pressure on the nerves in the area of the bleed. Damage to nerves can cause numbness and decreased ability to use the injured limb.
Christmas disease varies from mild to severe, but mild cases are more common. The severity depends on the degree of deficiency of factor IX. Hemophilia B symptoms are similar to those of hemophilia A, including numerous large and deep bruises and prolonged bleeding.
Some of the most problematic and frequent bleeds occur into the joints, particularly the knees and elbows. Repeated bleeding into joints can result in scarring within the joints and permanent deformities. Individuals may develop arthritis in joints that have suffered continued irritation from the presence of blood. Mouth injuries can result in compression of the airway, which interrupts breathing and can be life-threatening. A blow to the head, which might be totally insignificant in a normal child, can result in bleeding into the skull and brain. Because the skull has no room for expansion, the hemophiliac is at risk for brain damage due to blood taking up space and exerting pressure on the delicate brain tissue.
People with hemophilia are at very high risk of severe, heavy, uncontrollable bleeding (hemorrhage) from injuries such as motor vehicle accidents and also from surgery.
Some other rare clotting disorders such as von Willebrand's disease present similar symptoms but are not usually called hemophilia.
When to call the doctor
Hemophilia is usually discovered when an injury initiates bleeding and the bleeding will not stop. In very young children, spontaneous musculoskeletal bleeding may occur around the time the child begins to walk; these episodes may be the first sign of hemophilia. In some children, a simple surgical procedure, such as a tooth extraction or injection, may present with uncontrolled bleeding. Any signs of deep bruises or the presence of prolonged bleeding after a bump or an injury that breaks the skin should be reported to a physician or emergency service immediately. Bleeding under the skin (hematoma), which looks like a severe bruise, should also be reported and medical care sought immediately.
Diagnosis
Various diagnostic tests are available to measure, under carefully controlled conditions, the length of time it takes to produce certain components of the final fibrin clot. The activated partial thromboplastin time (APTT) is performed and will typically be prolonged while a prothrombin time (PT) will likely be normal. Factor assays, measurement methods performed by the clinical laboratory, can determine the percentage of factors VIII and IX present compared to normal percentages. This information helps to confirm a diagnosis of hemophilia and identifies the type and severity of hemophilia present.
Families with a history of hemophilia can also have tests done during a pregnancy to determine whether the fetus will have hemophilia. Chorionic villous sampling is a test that examines proteins for deficiencies or defects that are characteristic of hemophilia. The test can be performed at 10 to 14 weeks; test performance is associated with a 1 percent risk of miscarriage. Amniocentesis is a method of withdrawing amniotic fluid from the placenta to allow examination of fetal cell DNA shed into the amniotic fluid, helping to identify genetic mutations. Amniocentesis can be performed at 15 to 18 weeks gestation and is associated with a one in 200 risk of miscarriage.
Treatment
The treatment of hemophilia involves replacing or supplementing the deficient coagulation factors. Various preparations of factors VIII and IX are available to replace missing factors as needed. Cryoprecipitate, for example, is a single- or multiple-donor human plasma preparation rich in coagulation factors; it is made available as a frozen concentrate. Fresh frozen plasma is a single-donor preparation of factor-rich plasma; it is used primarily for replacing factor XI in individuals with hemophilia C. Concentrated factor preparations may be obtained from a single donor, by pooling the donations of as many as thousands of donors, or by laboratory creation through highly advanced genetic techniques. These preparations are administered directly into the individual's veins (intravenous administration).
The frequency of treatment with coagulation factors depends on the severity of the individual's disease. Relatively mild disease will only require treatment in the event of injury, or to prepare for scheduled surgical or dental procedures. More severe disease will require regular treatment to avoid spontaneous bleeding.
Appropriate treatment of hemophilia can decrease suffering and be lifesaving in the presence of hemorrhage. Complications associated with treatment, however, can also be quite serious. About 20 percent of all individuals with hemophilia A begin to produce antibodies in their blood against the specific factor protein; the presence of antibodies may then rapidly destroy infused factor VIII. The presence of such antibodies may greatly hamper efforts to prevent or stop a major hemorrhage.
Individuals who receive coagulation factors prepared from pooled donor blood were once at risk for serious infections that could be passed through the infusion of human blood products, such as the hepatitis virus and HIV. Concern has also been raised about the possibility
Molecular biological techniques have introduced gene therapies as new treatment possibilities for hemophilia. Gene therapy involves sophisticated methods of transferring new genes to hemophiliacs, correcting deficiencies or defects in the clotting mechanism. These methods are being researched in the early 2000s.
Prognosis
Variations in the type and severity of hemophilia makes it difficult to generalize a prognosis, however, for individuals with mild hemophilia, the prognosis is quite good. Those with more severe hemophilia can also live relatively normal lives with careful management and avoidance of injury. Many individuals achieve normal life expectancy. Without treatment of bleeding episodes, severe muscle and joint pain and eventually permanent damage can occur. Much depends upon the physical activity level of the individual and the possibility of accidental injuries or surgeries required for other conditions, which cannot be predicted.
Prevention
Because of its genetic origins, hemophilia cannot be prevented in those born with the inherited defects or factor deficiencies. However, individuals who have a family history of hemophilia may benefit from genetic testing and counseling before deciding to have a baby.
The most important way for individuals with hemophilia to prevent complications of the disease is to avoid activities that may lead to injury. Those individuals who require dental work or any type of surgery may need to be pre-treated with an infusion of factor VIII to avoid hemorrhage. Hemophiliacs should also avoid medications or drugs that promote bleeding; aspirin is one such medication and many prescription drugs have anticoagulant properties.
Parental concerns
When a child has an inherited coagulation disorder such as hemophilia, parents will be concerned about the possibility of trauma or injury that may lead to potentially dangerous bleeding episodes. The watchfulness of parents along with effective management of hemophilia by physicians can help the child to lead a relatively normal life. Careful avoidance of injury is essential. Counseling is available to help children handle the psychosocial aspects of living with hemophilia. Education is available from public health organizations to help parents be informed about their child's condition.
KEY TERMS
Amplification —A process by which something is made larger. In clotting, only a very few chemicals are released by the initial injury; they trigger a cascade of chemical reactions which produces increasingly larger quantities of different chemicals, resulting in an appropriately-sized, strong fibrin clot.
Coagulation factors —Specific coagulation proteins in the blood required for clotting. Coagulation proteins are designated with roman numerals I through XIII.
Fibrin —The last step in the blood coagulation process. Fibrin forms strands that add bulk to a forming blood clot to hold it in place and help "plug" an injured blood vessel wall.
Hemorrhage —Severe, massive bleeding that is difficult to control. The bleeding may be internal or external.
Mutation —A permanent change in the genetic material that may alter a trait or characteristic of an individual, or manifest as disease. This change can be transmitted to offspring.
Platelet —A cell-like particle in the blood that plays an important role in blood clotting. Platelets are activated when an injury causes a blood vessel to break. They change shape from round to spiny, "sticking" to the broken vessel wall and to each other to begin the clotting process. In addition to physically plugging breaks in blood vessel walls, platelets also release chemicals that promote clotting.
Trauma —Serious physical injury. Also refers to a disastrous or life-threatening event that can cause severe emotional distress, including dissociative symptoms and disorders.
See also Coagulation disorders .
Resources
BOOKS
Britton, Beverly. Diseases and Disorders: Hemophilia. Farmington Hills, MI: Gale, 2003.
Khoury, Muin J., Wylie Burke, and Elizabeth J. Thomson, eds. Genetics and Public Health in the 21st Century: Using Genetic Information to Improve Health and Prevent Disease. New York: Oxford University Press, 2000.
McDougald, Monroe. Hemophilia Care in the New Millennium. Lancaster, UK: Kluwar Academic Publishers, 2001.
Rodriguez-Merchan, E. C., et al. Inhibitors in Patients with Hemophilia. Oxford, UK: Blackwell Publishing, 2002.
ORGANIZATIONS
National Hemophilia Foundation. 116 West 32nd St., 11th Floor, New York, NY 10001. Web site: http://www.hemophilia.org.
National Organization for Rare Disorders (NORD). PO Box 8923, New Fairfield, CT 06812–8923. Web site: http://www.rarediseases.org.
WEB SITES
"Hemostatis and Coagulation." The Merck Manual Online , 2003. Available online at http://www.merckcom/pubs/mmanual/section11/chapter131.131c.htm (accessed October, 22, 2004).
March of Dimes. Available online at http://www.modimes.org (accessed October 22, 2004).
L. Lee Culvert Jennifer F. Wilson, MS
Comment about this article, ask questions, or add new information about this topic: