Sickle cell anemia
Definition
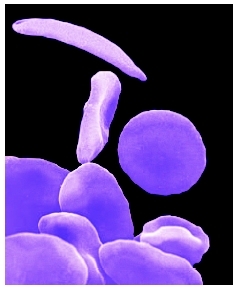
Sickle cell anemia, also called sickle cell disease (SS disease), is an inherited condition caused by having abnormal hemoglobin, the protein that carries oxygen in the blood. People with sickle cell anaemia have sickle hemoglobin (HbS) which is different from the normal hemoglobin (HbA).
Description
Children with sickle cell anemia produce two abnormal hemoglobin proteins (inheriting one from each parent), which makes their red blood cells easily destructible while giving them a sickle-like shape. Since the red blood cells do not have a normal shape, their circulation in the small blood vessels is impaired as well as the function of the abnormal hemoglobin (HbS) which can no longer carry oxygen with maximum efficiency.
Transmission
Sickle cell anemia is usually inherited from parents who are carriers, who have the sickle cell trait—a milder form of sickle cell anemia, or one abnormal hemoglobin.
Demographics
Sickle cell anemia and sickle cell trait are found mainly in people whose families come from Africa, the Caribbean, the Eastern Mediterranean, Middle East, and Asia. In the United States, sickle cell anemia affects some 72,000 people. The families of most of the people affected come from Africa. The disease occurs in about one in every 600 African-American births and in one in every 1,000 to 1,400 Hispanic-American births. Some 2 million Americans carry the sickle cell trait and about one in 12 African Americans have the trait.
Causes and symptoms
Sickle cell anemia is caused by an error in the gene that signals the body how to make hemoglobin. The defective gene tells the body to make the abnormal hemoglobin HbS instead of the normal HbA, and this results in deformed red blood cells. The error in the hemoglobin gene is due to a genetic mutation that occurred many thousands of years ago in people living in Africa, the Mediterranean basin, the Middle East, and India. A deadly form of malaria was very common at that time, and research has shown that in areas where malaria was endemic, children who inherited one HbS gene and who, therefore, carried the sickle cell trait, had a survival advantage because, unlike the children who had normal HbA genes, they survived malaria. They grew up, had their own children, and passed on the gene for HbS.
Symptoms or complications associated with sickle cell anemia usually start after the age of four to six months and can include all or some of the following:
- anemia, caused by low amounts of red blood cells in the bloodstream, resulting in insufficient oxygen delivery to tissues and organs
- vaso-occlusive pain , meaning severe episodes of pain in the arms, legs, or back, due to impaired blood circulation in the blood vessels
- chest pain and fever with coughing
- dactylitis, or hand-foot syndrome, with painful swelling of the bones in the hands or feet of young children
- aplastic crises, during which the body stops making new red blood cells causing severe anemia usually following an infection with the parovirus B19, which causes fifth disease
- priapism, a painful and prolonged erection
- stroke , usually causing sudden weakness of one side of the body
- acute splenic sequestration, with pooling of blood causing a sudden enlargement of the spleen
- jaundice, a yellowing of the skin and white of the eyes
- frequent infection with certain bacteria, particularly pneumococcus and salmonella, which are due to auto-infarction of the spleen or death of the spleen due to poor blood flow
When to call the doctor
Children suffering from sickle anemia have episodes during which they suddenly become unwell or complain of severe abdominal or chest pain, headache , stiffness of the neck or drowsiness. Parents should know that a child having a sickle cell crisis requires urgent hospital treatment. They should also call a doctor if the child has a temperature above 101°F (38.5°C).
Diagnosis
The diagnosis of sickle cell anemia is established during the newborn screen testing that is performed in the nursery at time of birth. For children who are not tested, an electrophoresis test of the blood can detect the abnormal hemoglobin of sickle cell anemia. This test measures the speed at which a molecule moves in a gel and can detect abnormal hemoglobin HbS.
Treatment
Treatment usually includes frequent monitoring of red blood counts, antibiotics for infections, transfusions for aplastic crises and splenic sequestration when required, and oxygen as well as respiratory support for chest syndrome. Some patients with severe symptoms receive regular blood transfusions to prevent crises and/or other complications such as stroke and organ damage.
Children with sickle cell disorders are at risk of developing severe infections, and penicillin is usually prescribed to prevent dangerous pneumococcal infections.
Sickle cell pain can be managed with a variety of measures including the following:
- warmth, to increase the blood flow, by massaging and rubbing and by heat from hot water bottles and deep heat creams
- bandaging to support the painful region
- rest
- getting the child to relax, by deep breathing exercises and distracting the attention, and by other psychological methods
- use of pain-killing medicines ( analgesics )
Analgesics should only be given as recommended by the treating physician. The gentlest analgesic usually prescribed is paracetamol, given three times a day (62.5 mgm under 12 months; 125 mgm 1–4 years; 250 mgm 4–10 years; 500 mgm 10–14 years; and 1 gm 15 years upwards). The next gentlest is codeine phosphate, given four times a day, at 1–2 mgm for every kilogram of body weight.
Bone marrow transplantation has been shown to provide a cure for severely affected children with sickle cell disease, but the procedure is not entirely without risk. In addition, the marrow must come from a healthy matched sibling donor and only about 18 percent of children with sickle cell anemia are likely to have a matched sibling.
Alternative treatment
Research contributed a great deal about sickle cell anemia from 1970 to the early 2000s concerning what causes it, how it affects the patient, and how to treat it. Scientists were as of 2004 starting to be successful at developing drugs that prevent the symptoms of sickle cell anemia and procedures that they hope should eventually provide a cure. Drug research is focused on identifying drugs, such as hydroxyurea, that can increase the level of fetal hemoglobin in the blood. Fetal hemoglobin is a form of hemoglobin that all humans produce before birth, but most stop producing it after birth. It has been observed that some children with sickle cell anemia continue to produce large amounts of fetal hemoglobin after birth, and studies have shown that these children have less severe cases of the disease. Fetal hemoglobin seems to prevent "sickling" of red cells, and cells containing fetal hemoglobin tend to survive longer in the bloodstream. Butyrate, a substance widely used as a food additive, was also being investigated as of 2004 as an agent that may increase fetal hemoglobin production.
Nutritional concerns
Thirst and dehydration caused by not drinking enough, even if thirst is not felt, are known to trigger sickle pain. Parents should accordingly monitor fluid intake closely.
Children with sickle cell anemia are anemic to various degrees. Most of the time they feel quite well, but if the anemia gets worse, they may feel very tired. Folic acid , a vitamin found in fruit and vegetables, supports making blood and anemic children especially need it to prevent them from becoming run down.
Prognosis
Sickle cell anemia is an inherited disease and lasts a lifetime.
Prevention
Both sickle cell trait and sickle cell anemia are inherited. Therefore, parents can pass it to their offspring. It is important for parents to get tested. If one partner has sickle cell trait and the other does not, their children each have a 50 percent chance of having the sickle cell trait, and a 50 percent chance of having normal hemoglobin. If one parent has sickle cell trait it is extremely important that the other parent be tested. If both parents have sickle cell trait, there is a 25 percent chance that the child will have normal hemoglobin, a 50 percent chance that the child will have the sickle cell trait, and a 25 percent chance the child will have sickle cell disease. If both parents have sickle cell trait and want to know whether the unborn child has sickle cell anemia, testing can be performed as early as the tenth week of pregnancy. If the results are normal, the parents can be reassured. If the tests show that the baby will be affected, the parents can be better prepared and they can make an informed decision concerning the pregnancy.
Parental concerns
Parents should be aware that children with sickle cell anemia are also at increased risk of infection, especially from the Streptococcus pneumonia and H. influenzae bacteria.
Sickle cell anemia does not affect intelligence . Children with sickle cell disorders can almost always attend school and participate fully in normal activities. The child's teacher and the school principal should know of the diagnosis and understand the limitations sickle cell anemia can impose on a child, for instance the need for frequent drinks and easy access to the bathroom, and the triggering of pain by over-exertion or cold.
KEY TERMS
Acute splenic sequestration —Retention of blood in the spleen.
Anemia —A condition in which there is an abnormally low number of red blood cells in the bloodstream. It may be due to loss of blood, an increase in red blood cell destruction, or a decrease in red blood cell production. Major symptoms are paleness, shortness of breath, unusually fast or strong heart beats, and tiredness.
Aplastic anemia —A disorder in which the bone marrow greatly decreases or stops production of blood cells.
Bone marrow —The spongy tissue inside the large bones in the body that is responsible for making the red blood cells, most white blood cells, and platelets.
Dactylitis —Inflammation of the hands or feet.
Endemic —Natural to or characteristic of a particular place, population, or climate.
Hemoglobin —An iron-containing pigment of red blood cells composed of four amino acid chains (alpha, beta, gamma, delta) that delivers oxygen from the lungs to the cells of the body and carries carbon dioxide from the cells to the lungs.
Priapism —A painful, abnormally prolonged penile erection.
Sickle cell trait —Condition that occurs in people who have one of two possible genes responsible for the abnormal hemoglobin of sickle cell anemia. People with this trait may suffer milder symptoms of sickle cell anemia or may have no symptoms. Some scientists believe that the trait actually provides an advantage in tropical environments because the slightly altered shape of the blood cells cause a person to be more resistant to malaria.
See also Anemias .
Resources
BOOKS
Gillie, Oliver. Sickle Cell Disease (Just the Facts). Chicago: Heineman Library, 2004.
Gordon, Melanie A. Let's Talk about Sickle Cell Anemia. New York: Powerkids Press, 2000.
Platt, Alan F., and A. Sacerdote. Hope and Destiny: A Patient's and Parent's Guide to Sickle Cell Disease and Sickle Cell Trait. Roscoe, IL: Hilton Publishing Co., 2003.
PERIODICALS
Fullerton, H. J., et al. "Declining stroke rates in Californian children with sickle cell disease." Blood 104, no. 2 (July 2004): 336–39.
Quinn, C. T., et al. "Survival of children with sickle cell disease." Blood 103, no. 11 (June 2004): 423–27.
Steinberg, M. H. "Therapies to increase fetal hemoglobin in sickle cell disease." Current Hematology Reports 2, no. 2 (March 2003): 95–101.
Zimmerman, S. A., et al. "Sustained long-term hematologic efficacy of hydroxyurea at maximum tolerated dose in children with sickle cell disease." Blood 103, no. 6 (March 2004): 239–45.
ORGANIZATIONS
American Sickle Cell Anemia Association (ASCAA). 10300Carnegie Avenue, Cleveland Clinic EEb18, Cleveland, OH 44106. Web site: http://www.ascaa.org.
Sickle Cell Society. 54 Station Road, London, NW10 4UA, UK. Web site: http://www.sicklecellsociety.org.
WEB SITES
"Sickle Cell Anemia." Medline Plus. Available online at http://www.nlm.nih.gov/medlineplus/sicklecellanemia.html (accessed November 14, 2004).
Monique Laberge, Ph.D.