Biliary atresia
Definition
Biliary atresia is the congenital failure of a fetus to develop an adequate pathway for bile to drain from the liver to the intestine.
Description
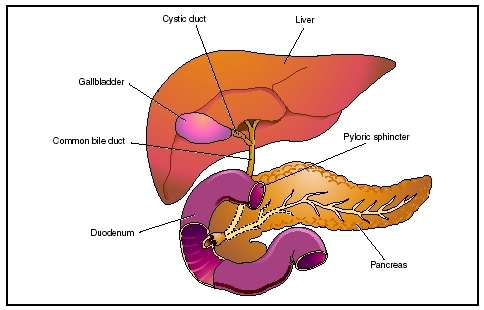
Biliary atresia is the congenital absence or closure of the ducts that drain bile from the liver. Bile is a liquid mixture of cholesterol, bile salts, and waste products, including bilirubin, which the liver excretes through thousands of tiny biliary ducts to the intestine, where the bile aids in the digestive process of dietary fats. These ducts merge into larger and larger channels, like streams flowing into rivers, until they all pour into a single duct that empties into the duodenum (first part of the small intestine). Between the liver and the duodenum this duct has a side channel connected to the gall bladder. The gall bladder stores bile and concentrates it, removing much of its water content. Then when food enters the stomach, the gall bladder contracts and empties its contents.
If bile cannot get out because the ducts are absent or blocked, it backs up into the liver (referred to as biliary stasis) and eventually into the rest of the body. The major pigment in bile is a chemical called bilirubin, which is yellow. Bilirubin is a breakdown product of hemoglobin (the red chemical in blood that carries oxygen). If the body accumulates an excess of bilirubin, it turns yellow (jaundiced). Bile also turns the stool brown; without it, stools are pale gray-, white- or fawn-colored. Bile trapped within the liver causes damage and scarring to the liver cells (cirrhosis of the liver). Scarring of the liver can cause portal hypertension (high blood pressure in the portal vein, which is the main vein carrying blood from the intestine to the liver). Portal hypertension may result in the development of fragile veins in the intestinal lining, stomach, or esophagus, which can bleed and require emergency medical attention.
Demographics
Biliary atresia is the most common lethal liver disease in children, occurring once every 10,000 to 15,000 live births. In the United States, approximately 300 cases of biliary atresia are diagnosed each year. Females are affected slightly more often than males. The incidence of biliary atresia is highest in Asian populations. The disorder also occurs in black infants at a rate approximately two times higher than that in white infants.
Causes and symptoms
The cause of biliary atresia is unknown. However, there are indications that viral infections or autoimmune mechanisms may be responsible for the development of biliary atresia. About 10 percent of children with biliary atresia also have other associated congenital defects in blood vessels, heart, spleen, or intestines.
The affected infant appears normal at birth and during the newborn period. After about two to three weeks, the infant develops jaundice . The infant has yellow eyes and skin and dark yellow or brown urine due to build-up of bilirubin, and the stools are probably light-colored. The child's abdomen begins to swell because of a firm, enlarged liver, and the infant gets progressively more ill. Weight loss and irritability will increase as the effects of jaundice increase. Some infants may develop intense itching (pruritis), which makes them even more uncomfortable. Nearly all untreated children die of liver failure within two years.
When to call the doctor
The doctor should be called if an infant older than two weeks of age exhibits jaundice or has other symptoms typical of biliary atresia.
If, after surgery for biliary atresia, an infant becomes jaundiced, has a high temperature for more than 24 hours, or if there is a change in the color of the stools or urine. Also after surgery, the infant may experience an abnormal collection of fluid in the abdomen, referred to as ascites, so the doctor should be consulted if the infant's stomach is distended.
If a child has black stools, pallor, or vomiting of blood due to the development of portal hypertension, emergency medical attention is required to treat the bleeding.
Diagnosis
The persistence or development of jaundice beyond the second week in a newborn who also has light-colored stools indicates obstruction to the flow of bile. An immediate evaluation that includes blood tests and imaging of the biliary system (through ultrasound, specialized x-ray techniques, or radioactive screens of the liver) are required to confirm the diagnosis. Other liver diseases that cause symptoms similar to biliary atresia must be ruled out through the testing process. In addition, in most cases, a liver biopsy or a surgical exploration of the infant's abdomen is necessary for a definitive diagnosis.
Treatment
Surgery is the only means to treat biliary atresia. The surgeon must create an adequate pathway for bile to escape the liver into the intestine. The altered anatomy of the biliary system is different in every case, calling upon the surgeon's skill and experience to select and execute the most effective among several options. If the obstruction is only between the gall bladder and the intestine, it is possible to attach a piece of intestine directly to the gall bladder.
If the upper biliary system is also inadequate, the surgeon will attach a piece of intestine directly to the liver using the Kasai procedure, named after Morio Kasai, the Japanese surgeon who developed the procedure. The tiny bile ducts in that part of the liver where the surgery is performed discharge their bile directly into the intestine, and the channels will gradually enlarge. A possible complication after the Kasai operation is an infection in the bile ducts (cholangitis). This infection must be treated immediately with intravenous antibiotics . If the child develops ascites (abnormal build-up of fluid in the abdomen), treatment consists of medications and alteration of the diet to maintain calorie intake but to reduce salt and fluid intake.
The operation is most successful in infants under the age of eight weeks. However, in many cases, liver damage may continue to occur, and without further intervention, cirrhosis of the liver and associated complications may develop. Continued problems often develop because there are also obstructed ducts within the liver that cannot be surgically treated. In these cases, liver transplantation is required. Improved techniques of liver transplantation, which allow transplantation in children of any age, and development of drugs that help overcome the problems of organ rejection offer significant hope to children with biliary atresia who are not successfully treated with surgical techniques.
Nutritional concerns
A low- or modified-fat diet with supplementary vitamins is often required after surgery, since the absorption of fats and vitamins can be impaired. Postoperative breastfeeding is encouraged whenever possible, as breast milk contains lipases and bile salts to aid in digestion. Infants who are formula-fed should use special formulas (Alimentum, Pregestimil) that contain chemicals to enhance digestion of dietary fats. Extra calories may also be required to help the infant gain weight. A dietary expert should be consulted to guide in the development of feeding requirements for an infant who has been treated surgically for biliary atresia.
Prognosis
Early diagnosis of biliary atresia is essential, for if left untreated, few children survive beyond the age of two years. If surgery is performed before the infant is two months old, success is much more likely, while after three months of age, the success rate is much poorer. Unfortunately for many infants, surgery is not a cure, and complications of cirrhosis of the liver may develop gradually, and the child eventually requires liver transplantation to avoid an early death. Transplantationasof2004achievesupto80to90percent one-year survival rates and promises to prevent the chronic disease that used to accompany earlier surgical procedures.
KEY TERMS
Cirrhosis —A chronic degenerative disease of the liver, in which normal cells are replaced by fibrous tissue and normal liver function is disrupted. The most common symptoms are mild jaundice, fluid collection in the tissues, mental confusion, and vomiting of blood. Cirrhosis is associated with portal hypertension and is a major risk factor for the later development of liver cancer. If left untreated, cirrhosis leads to liver failure.
Duodenum —The first of the three segments of the small intestine. The duodenum is about 10 in (25 cm) long and connects the stomach and the jejunum.
Hemoglobin —An iron-containing pigment of red blood cells composed of four amino acid chains (alpha, beta, gamma, delta) that delivers oxygen from the lungs to the cells of the body and carries carbon dioxide from the cells to the lungs.
Jaundice —A condition in which the skin and whites of the eyes take on a yellowish color due to an increase of bilirubin (a compound produced by the liver) in the blood. Also called icterus.
Prevention
Since the specific cause of this birth defect is unknown, there is no way known as of 2004 to prevent biliary atresia. However, it is not a hereditary condition.
Parental concerns
Parents of children with biliary atresia require help in coping with the strain of this chronic illness as well as the stress associated with waiting for a liver transplant. Parents may also feel guilty because they feel that they may have in some way contributed to the development of biliary atresia, although as of 2004, there is no known way to prevent the disease. The American Liver Foundation organizes and coordinates mutual help groups to provide emotional support for families, to make referrals to specialists as needed, and keep parents aware of research developments.
Resources
BOOKS
Kelly, Deirdre A., and Sheila Sherlock. Diseases of the Liver and Biliary System in Children , 2nd ed. Oxford, UK: Blackwell Publishers, 2004.
ORGANIZATIONS
American Liver Foundation. 75 Maiden Lane, Suite 603, New York, NY 10038. Web site: http://www.liverfoundation.org/.
Children's Liver Association for Support Services. 27023 McBean Parkway #126, Valencia, CA 91355. Web site: http://www.classkids.org/.
WEB SITES
Biliary Atresia Network. Available online at http://health.groups.yahoo.com/group/biliaryatresianetwork/ (accessed December 6, 2004).
"Patient Information about Biliary Atresia." Available online at http://www.pediatriconcall.com/fordoctor/DiseasesandCondition/Faqs/biliaryArt.asp (accessed December 6, 2004).
Judith Sims J. Ricker Polsdorfer, MD
Comment about this article, ask questions, or add new information about this topic: